

附件 1
甜叶菊多酚等 5种新食品原料
一、甜叶菊多酚
| 中文名称 | 甜叶菊多酚 |
| 英文名称 | Stevia polyphenols |
| 基本信息 | 来源:菊科甜菊属植物甜叶菊( Stevia rebaudianaBertoni)的叶 |
| 生产工艺简述 | 以甜叶菊的叶为原料,经乙醇提取、过滤、纯化、浓缩、干燥等工艺制成。 |
| 推荐食用量 | ≤500毫克 /天(以总多酚含量 40 g/100 g计,超过该含量的按照实际含量折算) |
| 其他需要说明的情况 | 1.使用范围和最大使用量:乳及乳制品(调制乳和风味发酵乳 0.5 g/kg,调制乳粉按照冲调后液体质量折算,干酪、再制干酪、干酪制品、炼乳按照生乳原料倍数折算),饮料类(液体饮料 ≤ 50 mL包装 5 g/kg, 51-500 mL包装 0.5 g/kg,固体饮料按照冲调后液体质量折算),果冻( 8 g/kg),可可制品、巧克力和巧克力制品(包括代可可脂巧克力及制品)( 8 g/kg),糖果( 25 g/kg),冷冻饮品(5 g/kg),酒类(2.5 g/kg),蜜饯( 5 g/kg)。 2.婴幼儿、孕妇和哺乳期妇女不宜食用,标签、说明书应当标注不适宜人群和食用限量。 3.质量规格和食品安全指标见附录。 |
1
附录
- 感官要求
- 感官要求应符合表 1的规定。表 1感官要求
- 理化指标
- 理化指标应符合表 2的规定。表 2理化指标
- 微生物指标
| 项目 | 要求 | 检测方法 |
| 色泽 | 棕色 | 取适量试样置于清洁、干燥的白瓷盘或烧杯中,在自然光线下,观察其色泽和状态,嗅其气味,品其滋味。 |
| 滋味 | 微甜,无异味 | |
| 气味 | 具有本品固有气味,无异味 | |
| 状态 | 粉末,无肉眼可见外来异物 |
| 项目 | 指标 | 检测方法 | |
| 总多酚(以没食子酸计),g/100 g | ≥ | 40.0 | 国家卫生健康委 2022年第 2号公告甘蔗多酚的总多酚测定方法 |
| 水分, g/100 g | ≤ | 6.0 | GB 5009.3 |
| 灰分, g/100 g | ≤ | 8.0 | GB 5009.4 |
| 铅( Pb),mg/kg | ≤ | 0.5 | GB 5009.12 |
| 总砷( As),mg/kg | ≤ | 0.5 | GB 5009.11 |
微生物指标应符合表 3的规定。表 3微生物指标
| 项目 | 指标 | 检测方法 | |
| 菌落总数, CFU/g | ≤ | 10000 | GB 4789.2 |
| 大肠菌群, CFU/g | ≤ | 10 | GB 4789.3 |
| 霉菌和酵母, CFU/g | ≤ | 50 | GB 4789.15 |
| 沙门氏菌, /25 g | 不得检出 | GB 4789.4 | |
| 金黄色葡萄球菌, /25 g | 不得检出 | GB 4789.10 |
二、柠檬香桃叶
| 中文名称 | 柠檬香桃叶 | |
|---|---|---|
| 英文名称 | Lemon myrtle leaf | |
| 基本信息 | 来源:桃金娘科檬香桃属植物柠檬香桃(Backhousiacitriodora F. Muell.)的叶 | |
| 生产工艺简述 | 以柠檬香桃的叶为原料,经采摘、筛选、清洗、干燥等工艺制成。 | |
| 其他需要说明的情况 | 1.婴幼儿、孕妇和哺乳期妇女不宜食用,标签、说明书应当标注不适宜人群。 2.食品安全指标须符合以下规定: | |
| 铅( Pb), mg/kg ≤ | 1.0 | |
| 镉( Cd), mg/kg ≤ | 0.5 | |
| 总砷( As), mg/kg ≤ | 0.5 | |
三、马基莓花色苷
| 中文名称 | 马基莓花色苷 |
| 英文名称 | Maqui berry anthocyanins |
| 基本信息 | 来源:杜英科酒果属植物马基莓( Aristotelia chilensis)的果实 |
| 生产工艺简述 | 以马基莓的果实为原料,经水提取、过滤、纯化、浓缩、干燥等工艺制成。 |
| 推荐食用量 | ≤900毫克 /天(以总花色苷含量 35 g/100 g计,超过该含量的按照实际含量折算) |
| 其他需要说明的情况 | 1.使用范围和最大使用量:乳及乳制品(调制乳和风味发酵乳 0.8 g/kg,乳粉及调制乳粉按照冲调后液体质量折算),饮料类(液体饮料 ≤50ml包装 8 g/kg,51-500ml包装 0.8 g/kg,固体饮料按照冲调后液体质量折算),果冻( 14 g/kg),可可制品、巧克力和巧克力制品(包括代可可脂巧克力及制品)( 14 g/kg),糖果( 40 g/kg),冷冻饮品( 8 g/kg),焙烤食品( 4 g/kg),酒类( 4 g/kg)。 2.婴幼儿、孕妇和哺乳期妇女不宜食用,标签、说明书应当标注不适宜人群和食用限量。 3.质量规格和食品安全指标见附录。 |
附录
- 感官要求
- 感官要求应符合表 1规定。表 1感官要求
- 理化指标
- 理化指标应符合表 2规定。表 2理化指标
- 微生物指标
| 项目 | 要求 | 检验方法 |
| 色泽 | 深紫色 | 取适量试样置于清洁、干燥的白瓷盘或烧杯中,在自然光线下,观察其色泽和状态,嗅其气味,品其滋味。 |
| 滋味 | 具有本品固有滋味,无异味 | |
| 气味 | 具有本品固有气味,无异味 | |
| 状态 | 粉末,无肉眼可见外来异物 |
| 项目 | 指标 | 检验方法 | |
| 总花色苷, g/100 g | ≥ | 35.0 | 附录 A |
| 水分, g/100 g | ≤ | 5.0 | GB 5009.3 |
| 灰分, g/100 g | ≤ | 5.0 | GB 5009.4 |
| 二乙烯苯, µg/kg | ≤ | 50.0 | 附录 B |
| 铅(Pb),mg/kg | ≤ | 0.1 | GB 5009.12 |
| 镉( Cd),mg/kg | ≤ | 0.1 | GB 5009.15 |
| 总汞( Hg),mg/kg | ≤ | 0.1 | GB 5009.17 |
| 总砷(As),mg/kg | ≤ | 0.5 | GB 5009.11 |
微生物指标应符合表 3的规定。表 3微生物指标
| 项目 | 指标 | 检验方法 | |
| 菌落总数, CFU/g | ≤ | 3000 | GB 4789.2 |
| 大肠菌群, CFU/g | ≤ | 10 | GB 4789.3 |
| 霉菌和酵母, CFU/g | ≤ | 100 | GB 4789.15 |
| 沙门氏菌, /25 g | 不得检出 | GB 4789.4 | |
| 金黄色葡萄球菌, /25 g | 不得检出 | GB 4789.10 |
附录 A总花色苷测定方法液相色谱法
- A.1原理
- A.2试剂和材料
- A.2.1甲醇,色谱纯。
- A.2.2乙腈,色谱纯。
- A.2.3 85%磷酸。
- A.2.4三氟乙酸,纯度≥ 99.5%。
- A.2.5矢车菊素 -3-O-葡萄糖苷标准品( CAS号: 7084-24-4),纯度≥ 96%。
- A.2.6 8.5%磷酸 -甲醇溶液:准确量取 85%磷酸 100 mL加入 800 mL的甲醇中,再用甲醇定容到 1000 mL,摇匀,即得。
- A.2.7 0.6%三氟乙酸水溶液:准确量取三氟乙酸 6 mL加入 800 mL的水,再用水定容到 1000 mL,摇匀,即得。
- A.2.8有机相微孔滤膜: 0.45 μm。
- A.3仪器和设备
- A.3.1分析天平:感量为 0.01 mg和 1 mg。
- A.3.2高效液相色谱仪:配紫外检测器。
- A.4分析步骤
- A.4.1标准溶液制备
- A.4.1.1标准储备液
- A.4.1.2标准工作液
- A.4.2试样溶液制备准确称取 100.0 mg样品并置于烧杯中,加入 10 mL的
8.5%磷酸 -甲醇溶液超声 2 min完全溶解,转移至 100 mL容量瓶中,冷却至室温,加入 80 mL8.5%磷酸 -甲醇溶液,超声 5 min,冷却至室温,然后再用 8.5%磷酸 -甲醇溶液定容至 100 mL。用 0.45 μm微孔滤膜过滤,即得试样溶液。
- A.4.3参考色谱条件
- a)色谱柱:耐酸性
- C18柱, 150 mm×4.6 mm,粒径 3.5 μm,或其他等效色谱柱;
- b)检测波长:
- 520 nm;
- c)流速:
- 0.7 mL/min;
- d)柱温:
- 30℃;
- e)进样量:
- 3 μL;
- f)流动相:流动相
- A:0.6%三氟乙酸水溶液;流动相 B:
乙腈。梯度洗脱程序见表 1。表 1梯度洗脱条件
| 时间( min) | 流动相 A(%) | 流动相 B(%) |
|---|---|---|
| 0 | 95 | 5 |
| 4 | 95 | 5 |
| 4.5 | 90 | 10 |
| 27 | 85 | 15 |
| 47 | 45 | 55 |
| 48 | 10 | 90 |
| 50 | 10 | 90 |
| 51 | 95 | 5 |
| 60 | 95 | 5 |
A.5测定
取标准工作液、试样溶液,依次注入高效液相色谱仪进行测定,按外标法计算总花色苷的含量。
A.6计算样品中 8种花色苷的含量按式( 1)计算:
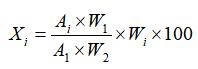
…………………(1)
式中:
Xi—i组分花色苷的含量,单位为克每百克( g/100 g);
i —a、b、c、d、e、f、g、h分别对应翠雀素 -3-O-桑布双糖苷 -5-O-葡萄糖苷、翠雀素 -3,5-O-二葡萄糖苷、矢车菊素 -3-O-桑布双糖苷 -5-O-葡萄糖苷、矢车菊素 -3,5-O-双葡萄糖苷、翠雀素 3-O-桑布双糖苷、翠雀素 -3-O-葡萄糖苷、矢车菊素 -3-O-桑布双糖苷、矢车菊素 -3-O-葡萄糖苷;
Wi—i组分与矢车菊素 -3-O-葡萄糖苷的分子量比值: 1.64(翠雀素 -3-O-桑布双糖苷 -5-O-葡萄糖苷)、 1.37(翠雀素 -3,5-O-二葡萄糖苷)、 1.61(矢车菊素 -3-O-桑布双糖苷 -5-O葡萄糖苷)、 1.33(矢车菊素 -3,5-O-双葡萄糖苷)、 1.31(翠雀素 -3-O-桑布双糖苷)、 1.03(翠雀素 -3-O-葡萄糖苷)、 1.27(矢车菊素 -3-O-桑布双糖苷)、 1.00(矢车菊素 -3-O-葡萄糖苷);
Ai—试样溶液色谱图中 i组分的峰面积;
A1—标准工作液色谱图中矢车菊素 -3-O-葡萄糖苷的峰面积;
W2—试样的质量,单位为克( g);
W1—矢车菊素 -3-O-葡萄糖苷标准品的质量,单位为克
(g)。
由式( 1)计算得到 8种组分的含量 Xa、Xb、Xc、Xd、 Xe、Xf、Xg、Xh,取各组分质量之和即为样品中总花色苷含量。
以重复性条件下获得的两次独立测定结果的算术平均值表示,结果保留小数点后一位有效数字。
A.7检出限和定量限
当样品取样量为 100.0 mg,定容体积为 100 mL时,本方法检出限为 14.8 mg/100 g,定量限为 49.3 mg/100 g。
- A.8精密度
- A.9液相色谱图

图 A.1矢车菊素 -3-O-葡萄糖苷标准溶液色谱图(0.1 mg/mL)
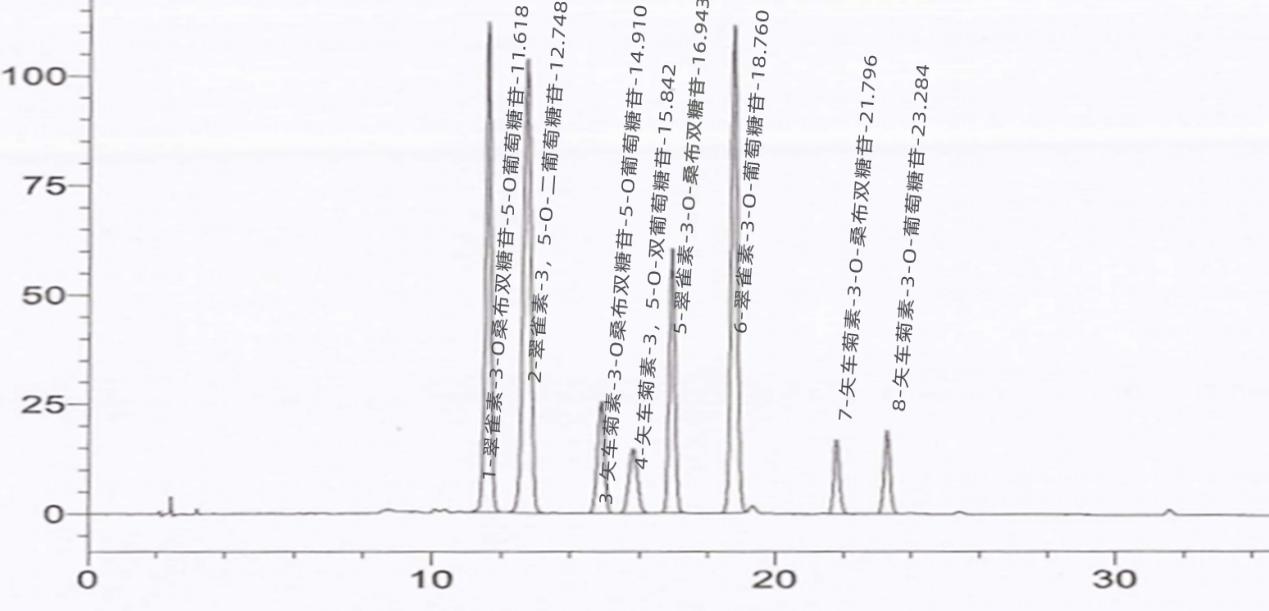
图 A.2试样溶液的参考色谱图( 1.0 mg/mL)
附录 B二乙烯苯残留量测定方法气相色谱 -质谱法
- B.1原理 试样经二氯甲烷提取,提取液经分散固相萃取净化后,用气相色谱 -质谱法测定,外标法定量。
- B.2试剂和材料 除非另有说明,本方法所用试剂均为分析纯,水为 GB/T 6682规定的一级水。
- B.2.1二氯甲烷,色谱纯。
- B.2.2 N-丙基乙二胺。
- B.2.3无水硫酸镁。
- B.2.4二乙烯苯标准品( CAS:1321-74-0)。
- B.3仪器和设备
- B.3.1气相色谱 -质谱联用仪:配电子轰击源。
- B.3.2分析天平:感量为 0.1 mg。
- B.3.3超声波清洗器。
- B.3.4离心机:转速≥ 4000 r/min。
- B.4分析步骤
- B.4.1标准溶液制备
- B.4.1.1标准储备液
准确称取 0.0430 g二乙烯苯于 25 mL容量瓶,用二氯甲烷溶解并定容至 25 mL,混匀,即得二乙烯苯浓度为 1000 mg/L的标准储备液( -10℃避光可保存 6个月)。
14
- B.4.1.2标准工作液
- B.4.1.3标准系列工作液
分别吸取标准储备液 0.00 mL、0.01 mL、0.10 mL、0.20 mL、0.50 mL、1.00 mL、2.00 mL于 100 mL容量瓶中,加二氯甲烷定容至 100 mL,混匀。此系列溶液二乙烯苯的浓度分别为 0.0 μg/L、1.0 μg/L、10.0 μg/L、20.0 μg/L、50.0 μg/L、
100.0 μg/L、200.0 μg/L。在参考色谱条件下,对标准系列工作液分别进样,以峰面积之和为纵坐标,标准工作液浓度为横坐标绘制标准工作曲线。
B.4.2试样溶液制备
准确称取样品 2g(精确到 0.01 g)于 25 mL具塞离心管,加入 10.0 mL二氯甲烷,称定重量。超声提取 20 min,冷却后称定重量,加二氯甲烷补足减失重量, 4000 r/min离心 2 min。
准确吸取 2.0 mL上清液于 15 mL具塞离心管,加入 150 mg N-丙基乙二胺和 900 mg无水硫酸镁,涡旋振荡 2 min, 4000 r/min离心 2 min,取上清液进样。
- B.4.3空白试样制备除不加试样外,按步骤 B.4.2制备空白试样。
- B.4.4参考条件
- B.4.4.1色谱参考条件
- a)色谱柱:含
- 6%氰丙基苯和 94%二甲基硅氧烷的毛细管柱(柱长 30 m,内径 0.25 mm,膜厚 1.4 μm),或其他等效色谱柱;
- c)柱温:初始温度
- 50℃,保持 2 min;以 20.0℃/min升温到 250℃,保持 3 min;
- d)载气:氦气,纯度≥ 99.999%,流速 1.0 mL/min;
- e)进样量:
- 1.0 μL;
- f)进样方式:脉冲分流进样;
- g)分流比:
- 5:1。
- B.4.4.2质谱参考条件
- a)电子轰击源:
- 70 eV;
- b)离子源温度:
- 230℃;
- c)四极杆温度:
- 150℃;
- d)监测方式:选择离子模式 (SIM);
- e)监测离子
- (m/z):定量离子: 130(100);定性离子: 128(35):115(30);
- B.5测定
- B.5.1定性测定
15 16
试样溶液按规定的条件进行测定,如果样液与标准溶液在二乙烯苯相同的保留时间处( ±0.5%)出现,则对其进行质谱确认,在扣除背景后的样品质谱图中,所有选择离子均出现,而且所选择的离子丰度与标准品的离子丰度比在允许误差范围内(见表 1),则可以判断样液中存在二乙烯苯残留。如果不能确证,应重新进样,改变扫描方式(如果有足够的灵敏度)或采用增加其他确证离子的方式来进一步确证。
表 1定性分析时相对离子丰度允许的最大偏差
| 相对离子丰度( %) | 允许偏差( %) |
|---|---|
| >50 | ±20 |
| 20-50 | ±25 |
| 10-20 | ±30 |
| ≤10 | ±50 |
B.5.2定量测定
根据样液中二乙烯苯残留量的情况,选定峰面积相近的标准溶液,标准溶液和样液中二乙烯苯响应值在仪器测定的线性范围内。在仪器最佳工作条件下,标准溶液和样液等体积穿插进样测定;测定二乙烯苯的峰面积,查标准曲线得到样液中二乙烯苯的浓度。在上述色谱质谱条件下,二乙烯苯标准溶液的色谱图见图 B.1。
17
B.6计算
样品中二乙烯苯的含量按式( 1)计算:
![]() ……………………(1)
……………………(1)
式中: X—样品中二乙烯苯的含量,单位为微克每千克(μg/kg); c—样液中二乙烯苯的浓度,单位为微克每升( μg/L); V—试样溶液体积,单位为毫升( mL); m—试样的质量,单位为克( g)。 以重复性条件下获得的两次独立测定结果的算术平均
值表示,结果保留小数点后一位有效数字。
- B.7检出限和定量限
- B.8精密度
- B.9色谱图
18
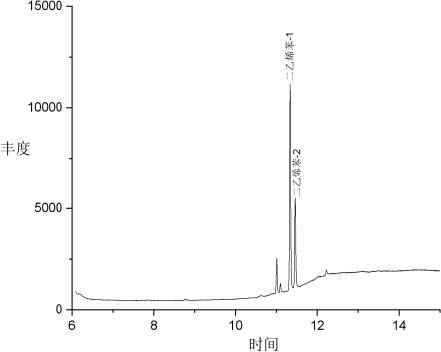
图 B.1二乙烯苯标准溶液选择离子质量色谱图
(10.0 μg/mL)
19
四、小麦极性脂质
| 中文名称 | 小麦极性脂质 |
| 英文名称 | Wheat polar lipids |
| 生产工艺简述 | 以小麦粉为原料,经乙醇提取、丙酮沉淀、分离、干燥、粉碎等工艺制成。 |
| 推荐食用量 | ≤30毫克 /天(以双半乳糖甘油二酯含量 40 g/100 g计,超过该含量的按照实际含量折算) |
| 其他需要说明的情况 | 1.使用范围和最大使用量:饮料类(液体饮料 ≤ 50 mL包装 0.6 g/kg,51-500 mL包装 0.06 g/kg,固体饮料按照冲调后液体质量折算)。 2.婴幼儿、孕妇和哺乳期妇女不宜食用,标签、说明书应当标注不适宜人群和食用限量。 3.质量规格和食品安全指标见附录。 |
20
附录
- 感官要求
- 感官要求应符合表 1的规定。表 1感官要求
- 理化指标
| 项目 | 要求 | 检测方法 |
| 色泽 | 米色至浅黄色 | 取适量试样置于清洁、干燥的白瓷盘或烧杯中,在自然光线下,观察其色泽和状态,嗅其气味,品其滋味。 |
| 滋味 | 具有本品固有滋味,无异味 | |
| 气味 | 具有本品固有气味,无异味 | |
| 状态 | 粉末,无肉眼可见外来异物 |
理化指标应符合表 2的规定。表 2理化指标
| 项目 | 指标 | 检测方法 | |
| 总糖脂, g/100 g | ≥ | 60.0 | 附录 A |
| 双半乳糖甘油二酯, g/100 g | ≥ | 40.0 | 附录 B |
| 酸价( KOH),mg/g | ≤ | 15.0 | GB 5009.229 |
| 过氧化值, mmol/kg | ≤ | 5.0 | GB 5009.227 |
| 水分, g/100 g | ≤ | 5.0 | GB 5009.3 |
| 铅(Pb),mg/kg | ≤ | 0.08 | GB 5009.12 |
| 镉(Cd),mg/kg | ≤ | 0.1 | GB 5009.15 |
| 总汞(Hg),mg/kg | ≤ | 0.1 | GB 5009.17 |
| 总砷(As),mg/kg | ≤ | 0.1 | GB 5009.11 |
| 溶剂残留, mg/kg | ≤ | 20.0 | GB 5009.262 |
21
3.微生物指标
微生物指标应符合表 3的规定。表 3微生物指标
| 项目 | 指标 | 检测方法 |
| 菌落总数, CFU/g ≤ | 100 | GB 4789.2 |
| 大肠埃希氏菌, MPN/g ≤ | 0.3 | GB 4789.38 |
| 霉菌和酵母, CFU/g ≤ | 100 | GB 4789.15 |
| 沙门氏菌, /25 g | 不得检出 | GB 4789.4 |
| 金黄色葡萄球菌, /25 g | 不得检出 | GB 4789.10 |
| 单核细胞增生李斯特氏菌, /25 g | 不得检出 | GB 4789.30 |
22
附录 A总糖脂测定方法分光光度法
- A.1原理
- A.2试剂和材料
- A.2.1蒽酮。
- A.2.2 98%浓硫酸。
- A.2.3葡萄糖标准品( CAS号: 50-99-7),纯度≥ 99%。
- A.3仪器和设备
- A.3.1分光光度计。
- A.3.2分析天平:感量为 0.1 mg。
- A.3.3恒温干燥箱。
- A.3.4恒温水浴锅。
- A.4分析步骤
- A.4.1蒽酮溶液制备
- A.4.2标准溶液制备
- A.4.2.1标准储备液
称取经 96℃±2℃干燥 2h的 0.5 g(精确到 0.0001 g)葡萄糖标准品,用水溶解并定容至 100 mL,得到 5.0 mg/mL葡
23
萄糖标准储备液( 4℃密封、避光可保存 1个月)。
A.4.2.2标准工作液
分别准确移取 0.0 mL、0.5 mL、1.0 mL、1.5 mL、2.0 mL葡萄糖标准储备液( A.4.2.1)于 100 mL容量瓶中,用水定容至 100 mL,配制成浓度为 0.000 g/L、0.025 g/L、0.050 g/L、
0.075 g/L、0.100 g/L的标准系列工作液。
- A.4.2.3标准曲线的制作
- A.4.3空白试验
- A.4.4试样溶液制备
- A.5计算
24
样品中葡萄糖当量浓度按式( 1)计算:
![]() ……………………( 1)
……………………( 1)
式中: x—样品中葡萄糖当量浓度,单位为克每百克( g/100 g); c—由标准曲线上查得的葡萄糖浓度,单位为克每升
(g/L); m—试样的质量,单位为克( g);
0.1—试样的定容量,单位为升( L); 100—试样重量以每百克计算的换算系数。 样品中总糖脂的含量按式( 2)计算:
![]() ……………………( 2)
……………………( 2)
式中: p—样品中总糖脂的含量,单位为克每百克( g/100 g); x—样品中葡萄糖当量浓度,单位为克每百克( g/100 g); 4—糖脂平均分子量( 720 g/mol)与葡萄糖分子量( 180
g/mol)之间的换算系数。以重复性条件下获得的两次独立测定结果的算术平均值表示,结果保留至小数点后一位有效数字。
- A.6检出限和定量限
- A.7精密度
25
在重复性条件下获得的两次独立测试结果的绝对差值不超过算术平均值的 10%。
26
附录 B双半乳糖甘油二酯测定方法液相色谱法
- B.1原理
- B.2试剂和材料
- B.2.1氯仿,色谱纯。
- B.2.2甲醇,色谱纯。
- B.2.3甲酸。
- B.2.4稀释液,氯仿 :甲醇 =3:1(v:v)。
- B.2.5双半乳糖甘油二酯( DGDG)标准品( CAS号: 92457-02-8),纯度≥ 98%。
- B.2.6有机相微孔滤膜: 0.45 μm。
- B.3仪器和设备
- B.3.1高效液相色谱仪:配蒸发光散射检测器。
- B.3.2超声波清洗器。
- B.3.3分析天平:感量为 0.1 mg。
- B.4分析步骤
- B.4.1标准溶液制备
- B.4.1.1标准储备液准确称取 12.5 mg DGDG标准品于 5 mL容量瓶中,加
27
入 2.5 mL稀释液溶解并定容至 5 mL,摇匀,即得 2.5 mg/mL的 DGDG标准储备液( 4℃密封、避光可保存 1个月)。
B.4.1.2标准工作液分别准确移取 0.12 mL、0.16 mL、0.20 mL、0.24 mL、
- mL的 DGDG标准储备液于 1 mL容量瓶中,用稀释液定容至 1 mL,得到浓度为 0.3 mg/mL,0.4 mg/mL,0.5 mg/mL,
- mg/mL,0.7 mg/mL的标准系列工作液。各溶液均超声 10 s,摇匀 5 min后备用。
- B.4.1.3标准曲线的制作
- B.4.2试样溶液制备
- B.4.3参考色谱条件
- a)色谱柱:
- C18柱, 250 mm×4.6 mm,粒径 5 µm,或其他等效色谱柱;
- b)流速:
- 1 mL/min;
- c)柱温:
- 40℃;
- d)进样量:
- 10 μL;
- e)流动相:流动相
- A:氯仿;流动相 B:甲醇 /水=95:5(v:v),
28
按最终体积加入 0.5%甲酸。梯度洗脱程序见表 1。表 1梯度洗脱程序
| 时间( min) | 流动相 A(%) | 流动相 B(%) |
|---|---|---|
| 0 | 99 | 1 |
| 14 | 76 | 24 |
| 15 | 20 | 80 |
| 20 | 20 | 80 |
| 21 | 99 | 1 |
| 30 | 99 | 1 |
B.5计算样品中 DGDG含量按式( 1)计算:
=10(−) ×� ×100………………(1)式中: X—样品中 DGDG含量,单位为克每百克( g/100 g); A—试样溶液中 DGDG的峰面积; a—DGDG标准曲线的斜率; b—DGDG标准曲线的截距; V—试样定容量,单位为毫升( mL); W—试样重量,单位为毫克( mg); 100—试样重量以每百克计算的换算系数。以重复性条件下获得的两次独立测定结果的算术平均值表示,结果保留至小数点后一位有效数字。
29
- B.6检出限和定量限
- B.7精密度
- B.8液相色谱图
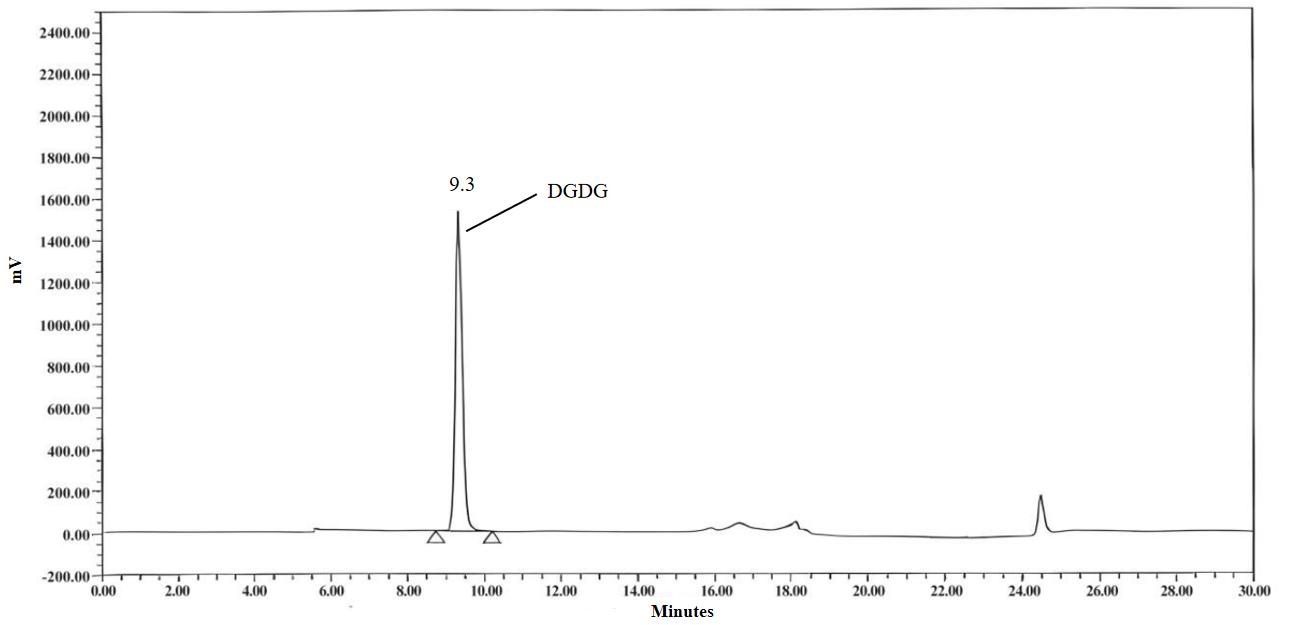
图 B.1 DGDG标准溶液色谱图( 0.5 mg/mL)
30
五、β-羟基-β-甲基丁酸钙
| 中文名称 | β-羟基 -β-甲基丁酸钙 | |
|---|---|---|
| 英文名称 | Calcium β-hydroxy-β-methyl butyrate (CaHMB) | |
| 基本信息 | 结构式:分子式: C10H18O6Ca∙H2O分子量: 292 | |
| 生产工艺简述 | 以次氯酸钠、二丙酮醇、盐酸、乙酸乙酯、乙醇、氢氧化钙为主要原料,经氧化合成、酸化、萃取、中和反应、离心、干燥等工艺制成。 | |
| 推荐食用量 | ≤6克/天 | |
| 质量要求 | 性状 | 白色粉末 |
| β-羟基 -β-甲基丁酸, g/100 g | 77-82(检测方法见附录 A) | |
| 钙, g/100 g | 12-16 | |
| 水分, g/100 g | 5.0-7.5 | |
| 2-羟基 -3-甲基 -2-丁烯酸, % ≤ | 8.0(检测方法见附录 B) | |
| 2,3-二羟基 -3-甲基丁酸, % ≤ | 4.5(检测方法见附录 B) | |
| 其他需要说明的情况 | 1.使用范围:饮料、乳及乳制品、可可制品、巧克力及巧克力制品、糖果、烘焙食品、运动营养食品、特殊医学用途配方食品。 | |
| 2.婴幼儿、儿童、孕妇和哺乳期妇女不宜食用,标签、说明书应当标注不适宜人群和食用限量。 3.食品安全指标须符合以下规定: | ||
|---|---|---|
| 铅(Pb),mg/kg ≤ | 1.0 | |
| 总砷(As),mg/kg ≤ | 1.0 | |
| 三氯甲烷, mg/kg ≤ | 1.0(检测方法见附录 C) | |
| 菌落总数, CFU/g ≤ | 1000 | |
| 大肠菌群, CFU/g ≤ | 10 | |
附录 A β-羟基 -β-甲基丁酸测定方法液相色谱法
- A.1原理
- A.2试剂和材料
- A.2.1磷酸二氢钾。
- A.2.2乙腈,色谱纯。
- A.2.3 85%磷酸。
- A.2.4氢氧化钾。
- A.2.5 36.0%-38.0%盐酸。
- A.2.6盐酸溶液( 0.1 mol/L):移取 8.3 mL盐酸,小心注入已加入适量水的 1000 mL容量瓶中,用水定容至 1000 mL,混匀。
- A.2.7氢氧化钾溶液( 1.0 mol/L):称取 56.0 g氢氧化钾于 1000 mL容量瓶,加适量水溶解,并用水定容至 1000 mL,混匀。
- A.2.8 β-羟基 -β-甲基丁酸标准品( CAS号: 625-08-1),纯度 ≥95%。
- A.2.9有机相微孔滤膜: 0.22 μm。
- A.2.10流动相 A:称取 2.60 g磷酸二氢钾至 1L烧杯中,加
- A.2.11流动相 B:分别量取 800 mL乙腈和 200 mL水,加入试剂瓶中,混合均匀。用微孔滤膜过滤后超声脱气,置于密闭容器中备用。
- A.3仪器和设备
- A.3.1高效液相色谱仪:配紫外检测器或二极管阵列检测器。
- A.3.2分析天平:感量为 0.1 mg。
- A.4分析步骤
- A.4.1标准曲线的制作
- A.4.1.1标准储备液
- A.4.1.2标准工作液
分别吸取 β-羟基 -β-甲基丁酸标准储备液 1.25 mL、2.50 mL、5.00 mL、7.50 mL和 10.00 mL于 10 mL容量瓶,用盐酸溶液定容至 10 mL,混合均匀,得到浓度分别为 0.225 mg/mL、0.450 mg/mL、0.900 mg/mL、1.350 mg/mL和 1.800
mg/mL的标准工作液。
- A.4.2试样溶液的制备
- A.4.3色谱参考条件
- a)色谱柱:
- C18色谱柱, 250 mm×4.6 mm,粒径 5 μm,或其他等效色谱柱;
- b)检测波长:
- 214 nm;
- c)流速:
- 0.5 mL/min;
- d)柱温:
- 20℃;
- e)进样量:
- 5 μL;
- f)流动相:流动相
- A:磷酸二氢钾( 20 mmol)/乙腈( 50 mL)混合溶液;流动相 B:乙腈水溶液( v:v=4:1)。梯度洗脱程序见表 1。
- A.4.4标准曲线的制作
- A.5测定
- A.6计算样品中 β-羟基 -β-甲基丁酸的含量按式( 1)计算:
| 时间( min) | 流动相 A(%) | 流动相 B(%) |
|---|---|---|
| 0 | 100 | 0 |
| 20 | 100 | 0 |
| 20.1 | 0 | 100 |
| 25 | 0 | 100 |
| 25.1 | 100 | 0 |
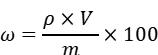 ……………(1) 式中: ω—样品中 β-羟基 -β-甲基丁酸的含量,单位为克每百克
……………(1) 式中: ω—样品中 β-羟基 -β-甲基丁酸的含量,单位为克每百克
(g/100 g); ρ—试样溶液中 β-羟基 -β-甲基丁酸的浓度,单位为毫克
每毫升( mg/mL); V—试样的定容体积,单位为毫升( mL); m—试样的质量,单位为毫克( mg);
100—换算系数。以重复性条件下获得的两次独立测定结果的算术平均值表示,结果保留至整数位。
- A.7检出限和定量限 当取样量为 0.14 g时,本方法检出限为 1.14 g/100 g,定量限为 3.79 g/100 g。
- A.8精密度 在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的 2%。
- A.9液相色谱图
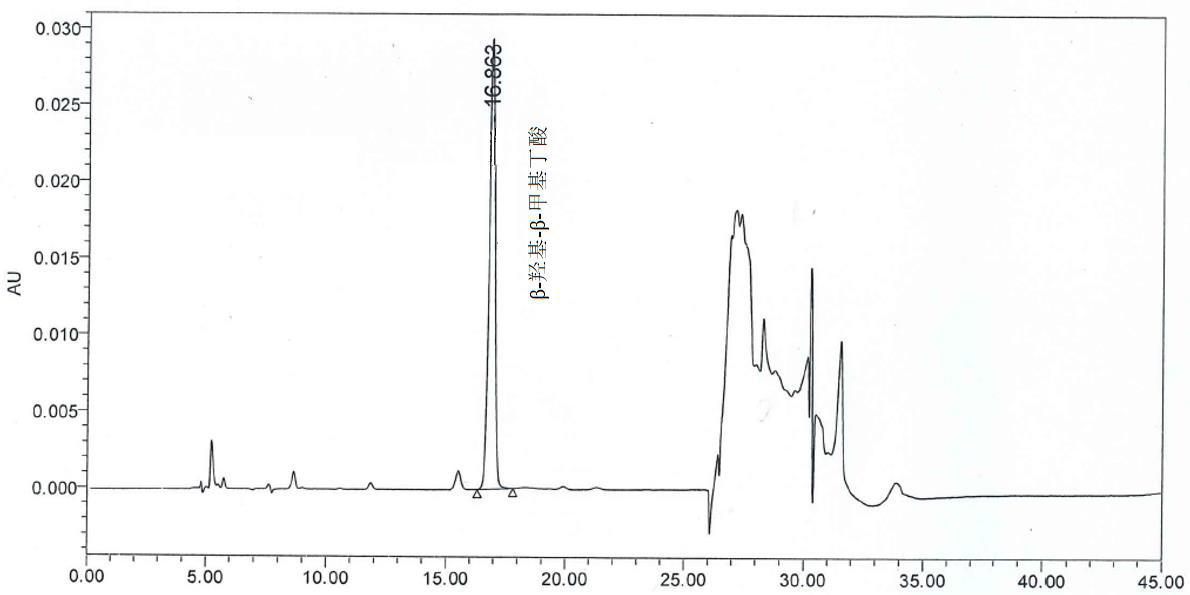
图 A.1 β-羟基 -β-甲基丁酸标准溶液的液相参考色谱图
(1.8 mg/mL)
附录 B 2,3-二羟基 -3-甲基丁酸和 2-羟基 -3-甲基 -2-丁烯酸测定方法液相色谱法
- B.1原理 试样经盐酸溶液溶解,经反相高效液相色谱分离,紫外吸收检测器测定。以试样溶液中 β-羟基 -β-甲基丁酸作为对照,采用相对保留时间定性试样溶液中的 2,3-二羟基 -3-甲基丁酸和 2-羟基 -3-甲基 -2-丁烯酸,面积归一化法定量。
- B.2试剂和材料 除非另有说明,本方法所用试剂均为分析纯,水为 GB/T 6682规定的一级水。
- B.2.1磷酸二氢钾。
- B.2.2乙腈,色谱纯。
- B.2.3 85%磷酸。
- B.2.4氢氧化钾。
- B.2.5 36.0%-38.0%盐酸。
- B.2.6盐酸溶液( 0.1 mol/L):移取 8.3 mL盐酸,小心注入已加入适量水的 1000 mL容量瓶中,用水定容至 1000 mL,混匀。
- B.2.7氢氧化钾溶液( 1.0 mol/L):称取 56.0 g氢氧化钾于 1000 mL容量瓶,加适量水溶解,并用水定容至 1000 mL,混匀。
- B.2.8 β-羟基 -β-甲基丁酸标准品( CAS号: 625-08-1),纯 度≥ 95%。
- B.2.9有机相微孔滤膜: 0.22 μm。
- B.2.10流动相 A:称取 2.60 g磷酸二氢钾至 1L烧杯中,加入 950 mL水,搅拌直至完全溶解,再加入 50 mL乙腈,搅匀。用约 0.25 g的 85%磷酸,调节 pH至 2.90±0.02;如果溶液 pH低于 2.88,再用氢氧化钾溶液( 1.0 mol/L)调节 pH至范围内。用微孔滤膜过滤后超声脱气备用。
- B.2.11流动相 B:分别量取 800 mL乙腈和 200 mL水,加入试剂瓶中,混合均匀。用微孔滤膜过滤后超声脱气,置于密闭容器中待用。
- B.3仪器和设备
- B.3.1高效液相色谱仪:配紫外检测器或二极管阵列检测器。
- B.3.2分析天平:感量为 0.1 mg。
- B.4分析步骤
- B.4.1标准溶液的制备 称取 β-羟基 -β-甲基丁酸标准品 0.18 g(精确至 0.0001 g)于烧杯中,用 0.1 mol/L盐酸溶液溶解后转移到 100 mL容量瓶中。用盐酸溶液溶解并定容至 100 mL,混合均匀,即得浓度为 1.8 mg/mL的标准储备液( 4℃可保存 4个月)。
- B.4.2试样溶液的制备 称取样品 0.56 g(精确到 0.0001 g),转移到 100 mL容量瓶中,加入盐酸溶液溶解并定容至 100 mL,混合均匀。
- B.4.3色谱参考条件
- a)色谱柱:
- C18色谱柱, 250 mm×4.6 mm,粒径 5 μm,或其他等效色谱柱;
- b)柱温:
- 20℃;
- c)流速:
- 0.5 mL/min;
- d)进样量:
- 5 μL;
- e)波长:
- 192 nm;
- f)流动相:流动相
- A:磷酸二氢钾( 20 mmol)/乙腈( 50 mL)混合溶液;流动相 B:乙腈水溶液( v:v=4:1)。梯度洗脱程序见表 1。
- B.5测定
| 时间( min) | 流动相 A(%) | 流动相 B(%) |
|---|---|---|
| 0 | 100 | 0 |
| 20 | 100 | 0 |
| 20.1 | 0 | 100 |
| 25 | 0 | 100 |
| 25.1 | 100 | 0 |
| 45 | 100 | 0 |
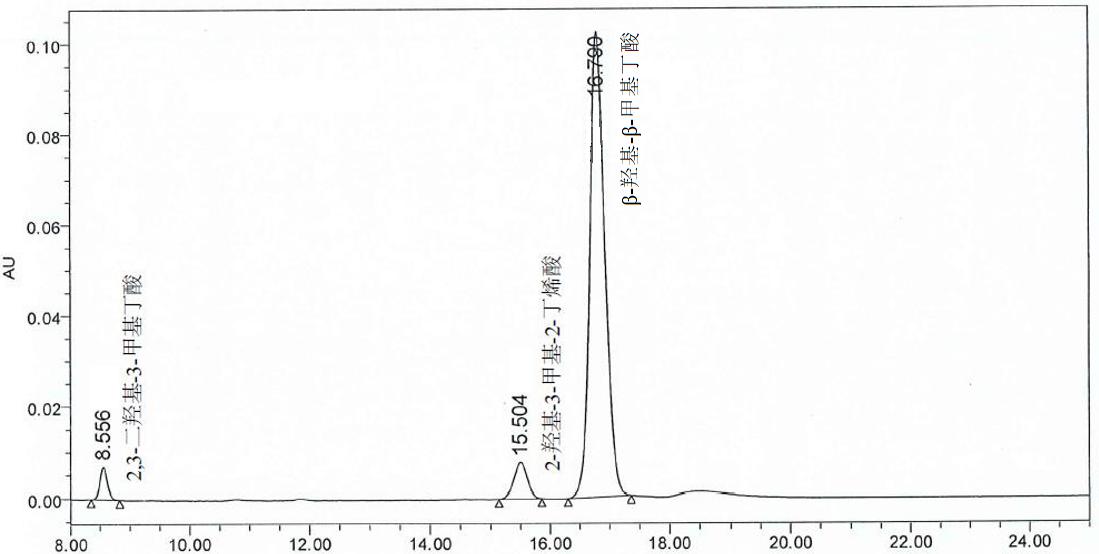
在规定色谱条件下,取盐酸溶液、 β-羟基 -β-甲基丁酸标准溶液和试样溶液各 5 μL,分别注入色谱仪进行测定。以试样溶液中 β-羟基 -β-甲基丁酸作为对照,采用相对保留时间(即试样溶液中某组分的保留时间与试样溶液中 β-羟基 -β甲基丁酸的保留时间的比值)定性 2,3-二羟基 -3-甲基丁酸和 2-羟基 -3-甲基 -2-丁烯酸(表 2)。
表2相对保留时间参考值
| 组分 | 相对保留时间 |
|---|---|
| 2,3-二羟基 -3-甲基丁酸 | 0.5 |
| 2-羟基 -3-甲基 -2-丁烯酸 | 0.9 |
| β-羟基 -β-甲基丁酸 | 1.0 |
B.6计算 样品中 2,3-二羟基 -3-甲基丁酸的含量按式( 1)计算:
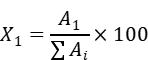 …………………(1)式中: X1—样品中 2,3-二羟基 -3-甲基丁酸的含量,单位为百分比( %); A1—试样溶液中 2,3-二羟基 -3-甲基丁酸的峰面积; ∑Ai—试样溶液中除溶剂峰之外的所有成分峰面积总和。以重复性条件下获得的两次独立测定结果的算术平均值表示,结果保留一位小数。样品中 2-羟基 -3-甲基 -2-丁烯酸的含量按式( 2)计算:
…………………(1)式中: X1—样品中 2,3-二羟基 -3-甲基丁酸的含量,单位为百分比( %); A1—试样溶液中 2,3-二羟基 -3-甲基丁酸的峰面积; ∑Ai—试样溶液中除溶剂峰之外的所有成分峰面积总和。以重复性条件下获得的两次独立测定结果的算术平均值表示,结果保留一位小数。样品中 2-羟基 -3-甲基 -2-丁烯酸的含量按式( 2)计算:
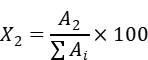 ……………………(2)式中: X2—样品中 2-羟基 -3-甲基 -2-丁烯酸的含量,单位为百分比( %);A2—试样溶液中 2-羟基 -3-甲基 -2-丁烯酸的峰面积; ∑Ai—试样溶液中除溶剂峰之外的所有成分峰面积总和。以重复性条件下获得的两次独立测定结果的算术平均值表示,结果保留一位小数。
……………………(2)式中: X2—样品中 2-羟基 -3-甲基 -2-丁烯酸的含量,单位为百分比( %);A2—试样溶液中 2-羟基 -3-甲基 -2-丁烯酸的峰面积; ∑Ai—试样溶液中除溶剂峰之外的所有成分峰面积总和。以重复性条件下获得的两次独立测定结果的算术平均值表示,结果保留一位小数。
- B.7检出限和定量限
- B.8精密度
- B.9液相色谱图
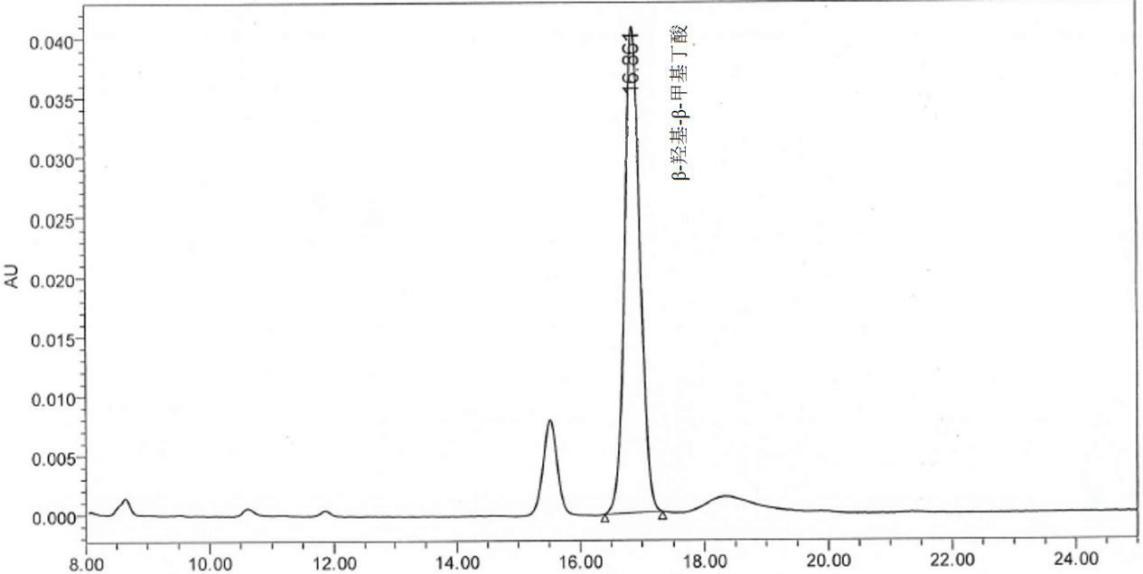
图 B.1 β-羟基 -β-甲基丁酸标准溶液的反相高效液相参考色谱图( 1.8 mg/mL)
时间(分钟)
图 B.2试样溶液中 2,3-二羟基 -3-甲基丁酸、 2-羟基 -3-甲基 -2丁烯酸和 β-羟基 -β-甲基丁酸的液相参考色谱图( 5.6 mg/mL)附录 C三氯甲烷的测定方法气相色谱法
- C.1原理
- C.2试剂和材料
- C.2.1甲醇,色谱纯。
- C.2.2三氯甲烷标准品( CAS:67-66-3),纯度≥ 99%。
- C.3仪器和设备
- C.3.1气相色谱仪:配有电子捕获检测器。
- C.3.2顶空瓶:容积 20-50 mL,使用前在 120℃烘烤 2h。
- C.3.3分析天平:感量为 0.1 mg。
- C.4分析步骤
- C.4.1标准储备液的制备
- C.4.2标准溶液的制备于200 mL容量瓶中加入 100 mL甲醇,再加入 1.0 mL的三
- C.4.3标准工作液的制备
- C.4.4试样溶液制备
- C.4.5色谱参考条件
- a)色谱柱:含
- 5%苯基亚芳基聚合物或 5%苯基 -甲基聚硅氧烷的弱极性毛细管柱(柱长 30 m,内径 0.32 mm,膜厚 0.25 μm),或其他等效色谱柱;
- b)汽化室温度:
- 200℃;
- c)柱温:
- 60℃;
- d)检测器温度:
- 200°C;
- e)载气流量:
- 2 mL/min;
- f)分流比:
- 10:1;
- g)尾吹气流量:
- 60 mL/min。
- C.4.6标准曲线的制作取6个200 mL容量瓶依次加入三氯甲烷标准工作液 0.00
mL、0.10 mL、0.50 mL、1.00 mL、2.00 mL和5.00 mL并用纯水定容至 200 mL,混匀。配制后三氯甲烷的浓度为 0.00 μg/L、
0.20 μg/L、1.00 μg/L、2.00 μg/L、4.00 μg/L、10.00 μg/L。分别准确移取标准系列工作液 10 mL于顶空瓶中,加盖密封,于70℃自动顶空平衡 20 min。取 1 mL注入气相色谱仪,以峰面积为纵坐标,浓度为横坐标绘制标准曲线。
- C.5测定
- C.6计算样品中三氯甲烷的含量按式( 1)计算。
![]() ……………………(1)
……………………(1)
式中: ρ—样品中三氯甲烷的含量,单位为毫克每千克( mg/kg); c—标准曲线上查得的试样溶液中三氯甲烷的含量,单位
为微克每升( μg/L); V—试样的定容体积,单位为毫升( mL); n—试样的稀释倍数; m—试样的质量,单位为克( g); 1000—单位换算系数。
以重复性条件下获得的两次独立测定结果的算术平均值表示,结果保留一位小数。
- C.7检出限和定量限
- C.8精密度
- C.9气相色谱图
图 C.1三氯甲烷标准溶液气相色谱图( 0.20μg/L)

